DISEASE OVERVIEW
MPS I is a rare, genetic, debilitating, multisystemic lysosomal storage disease1,2
Marked by heterogeneity, MPS I presents with a broad range of progressive manifestations and seemingly unrelated symptom combinations.3,4
Patients with MPS I either produce the enzyme in low amounts or not at all3
MPS I is a progressive disease caused by pathogenic variants in the IDUA gene leading to deficiency or complete lack of lysosomal enzyme IDUA. This deficiency in enzyme activity can result in widespread cellular, tissue, and organ dysfunction, which can be potentially life-threatening.3
MPS I can be severe and potentially life-threatening2,4

Without disease management, patients with the severe phenotype usually die within the first decade of life as a result of cardiorespiratory failure and progressive neurologic disease4

Without disease management, patients with the attenuated phenotype may have a normal or near-normal life span but may be burdened by considerable morbidity.4
Patients with Hurler-Scheie (H/S) syndrome may face premature death by the age of 30 years.4
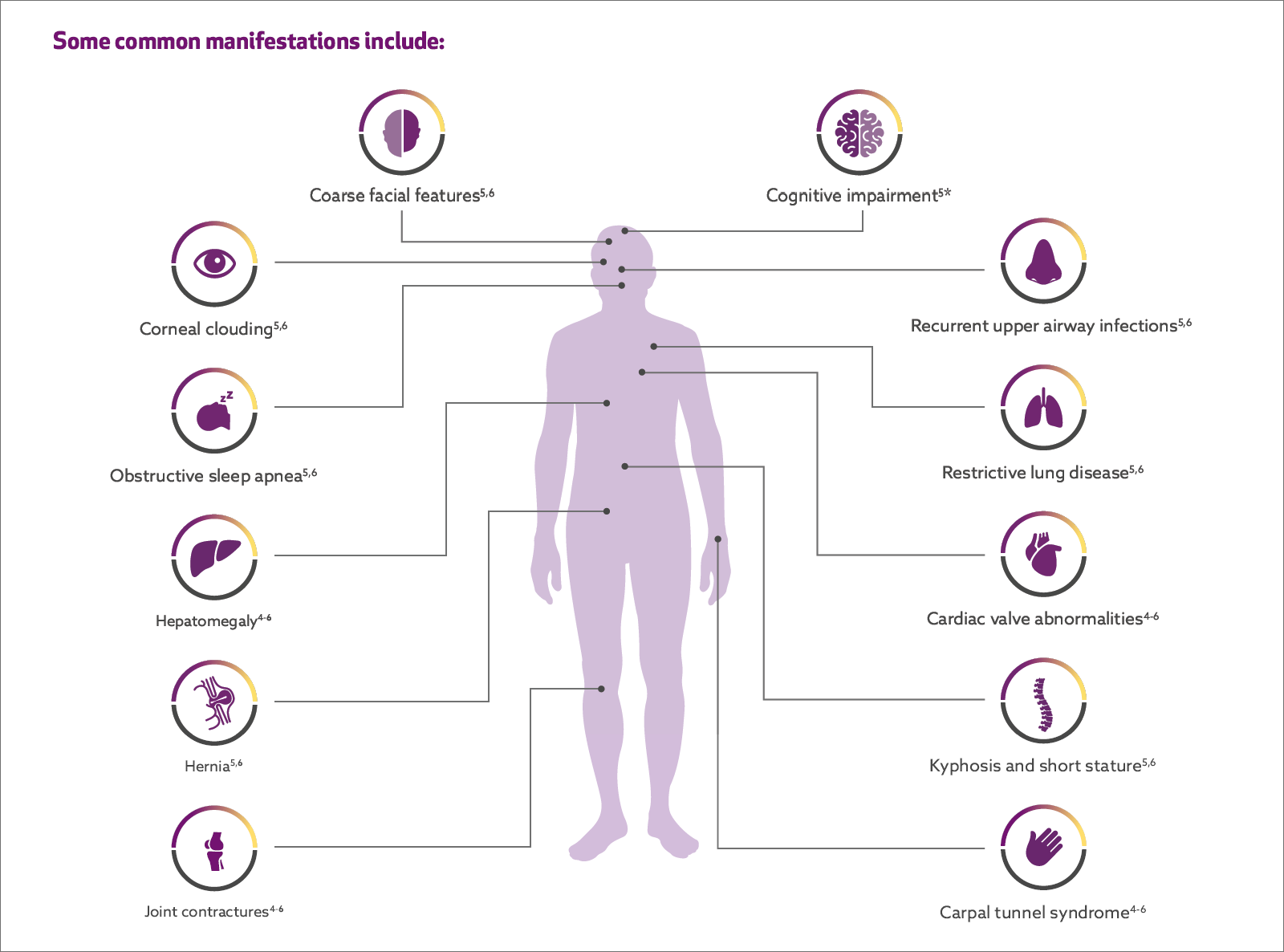
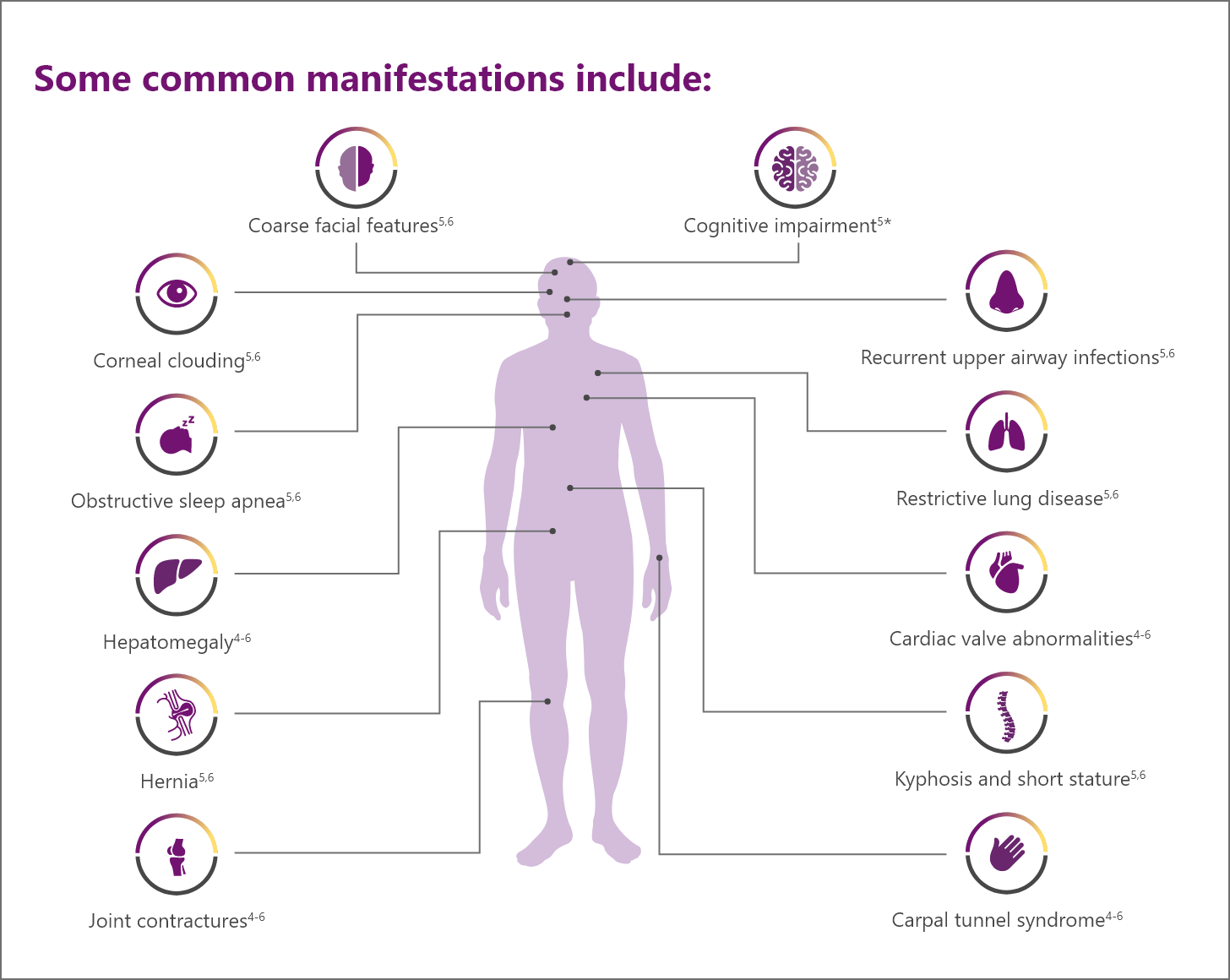
Patients with MPS I face a multitude of serious and oen debilitating multisystemic manifestations1,2
In MPS I, the IDUA enzyme is deficient or essentially absent, causing GAG substratesa to accumulate in the cells, which can lead to progressive damage to tissues and organs.
Progressive, multisystemic, potentially irreversible tissue damage can lead to loss of function, clinical deterioration, and a variety of manifestations.

Classification of MPS I3,4
MPS I is classified into 3 different syndromes based on the age of onset, severity of symptoms, and rate of disease progression.3,4
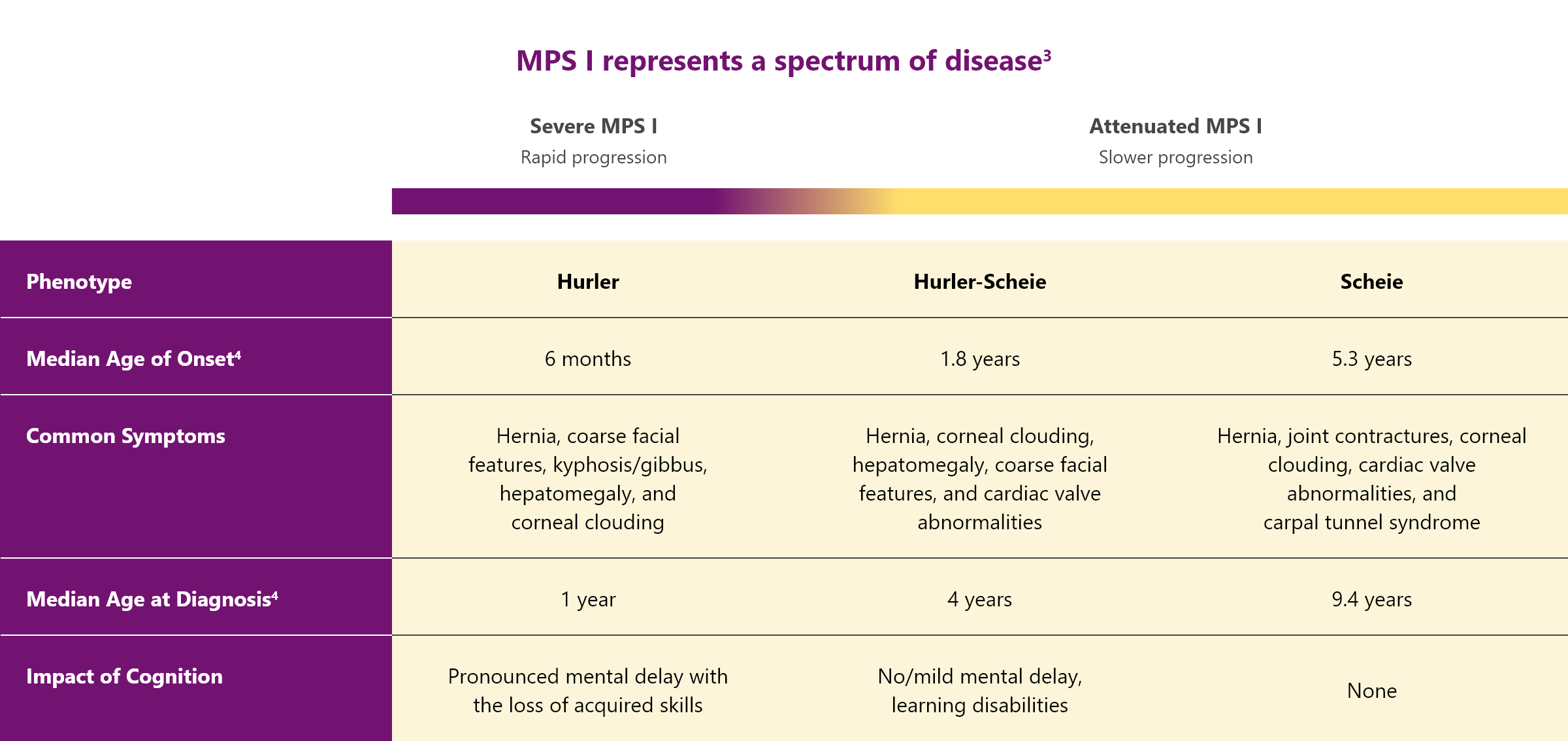
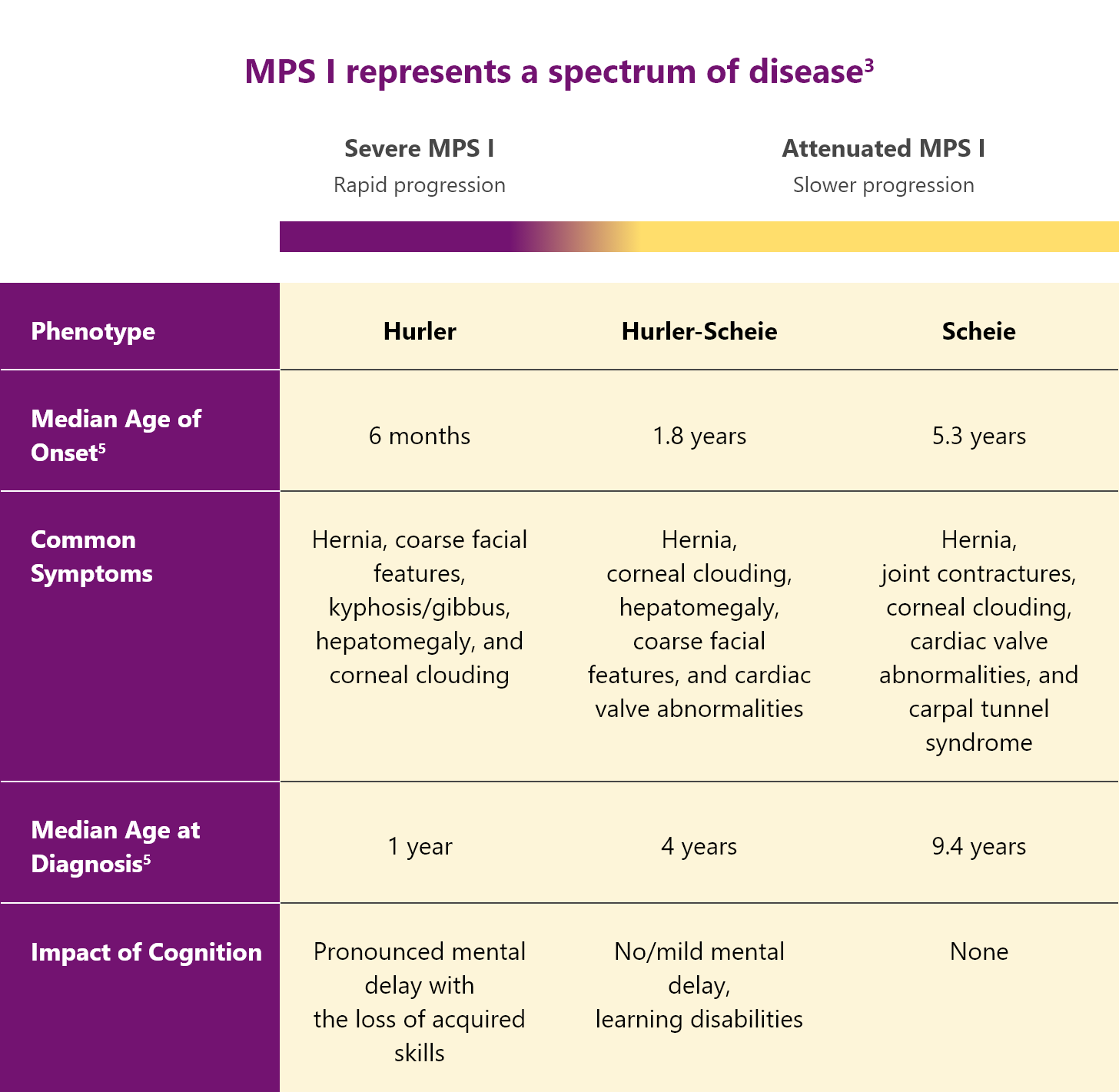
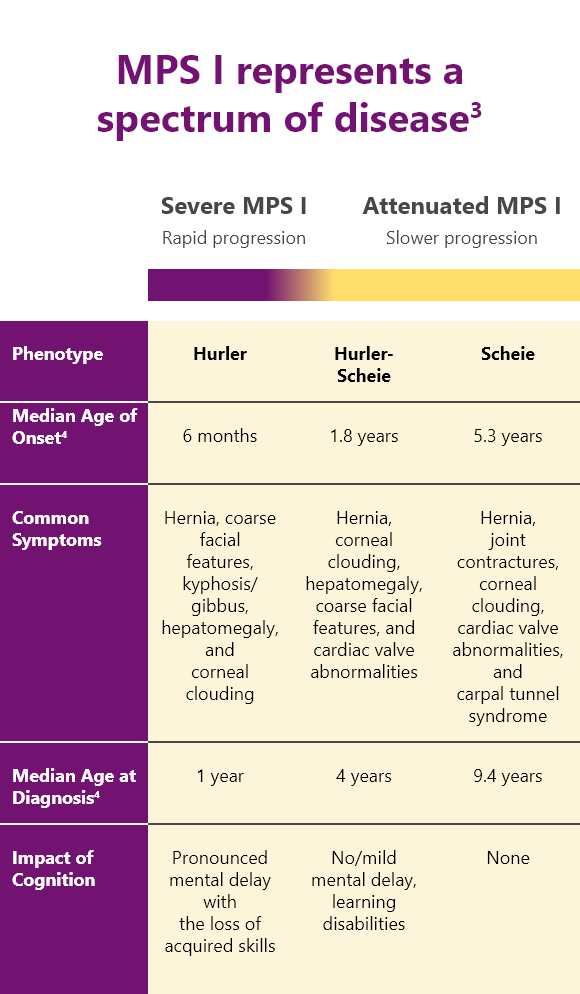
All patients with MPS I are at risk of significant, multisystemic disease
In severe MPS I
Patients with “severe” MPS I suffer from several debilitating symptoms including cognitive impairment that progress rapidly.3,4
In attenuated MPS I
Patients with “attenuated” MPS I have milder symptoms and a slower disease progression compared with Hurler syndrome. But they can still experience significant disease-related morbidity.3,4
CNS = central nervous system; GAG = glycosaminoglycan; H/S syndrome, Hurler-Scheie syndrome; IDUA = α-L-iduronidase; MPS I = mucopolysaccharidosis type I.
aThe GAGs that accumulate in MPS I are dermatan sulfate and heparan sulfate.
*Scheie form has no cognitive impairment/normal cognitive function.

References: 1. Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-L-iduronidase (laronidase). J Pediatr. 2004;144(5):581-588. 2. D’Aco K, Underhill L, Rangachari L, et al. Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry. Eur J Pediatr. 2012;171(6):911-919. 3. Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; 2019. Accessed July 20, 2022. https://ommbid.mhmedical.com/content.aspx?bookid=2709&ectionid=225544161. 4. Beck M, Arn P, Giugliani R, et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-765. 5. Muenzer J, Wraith JE, Clarke LA; International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. 6. Wraith JE. The first 5 years of clinical experience with laronidase enzyme replacement therapy for mucopolysaccharidosis I. Expert Opin Pharmacother. 2005;6(3):489-506.